

La fibrosi cistica (FC) è la più frequente malattia genetica letale al mondo e rappresenta un quesito dal punto di vista terapeutico fino ad oggi parzialmente irrisolto. Sebbene vi sia stato negli ultimi anni un progresso nella ricerca, le terapie rimangono sempre focalizzate sui sintomi, data l’impossibilità di curare il male alla radice, almeno fino ad oggi. Una speranza nuova giunge da una equipe composta in gran parte da medici e ricercatori italiani, che sembra aver aperto una nuova strada verso una cura definitiva.
Cos’è la fibrosi cistica
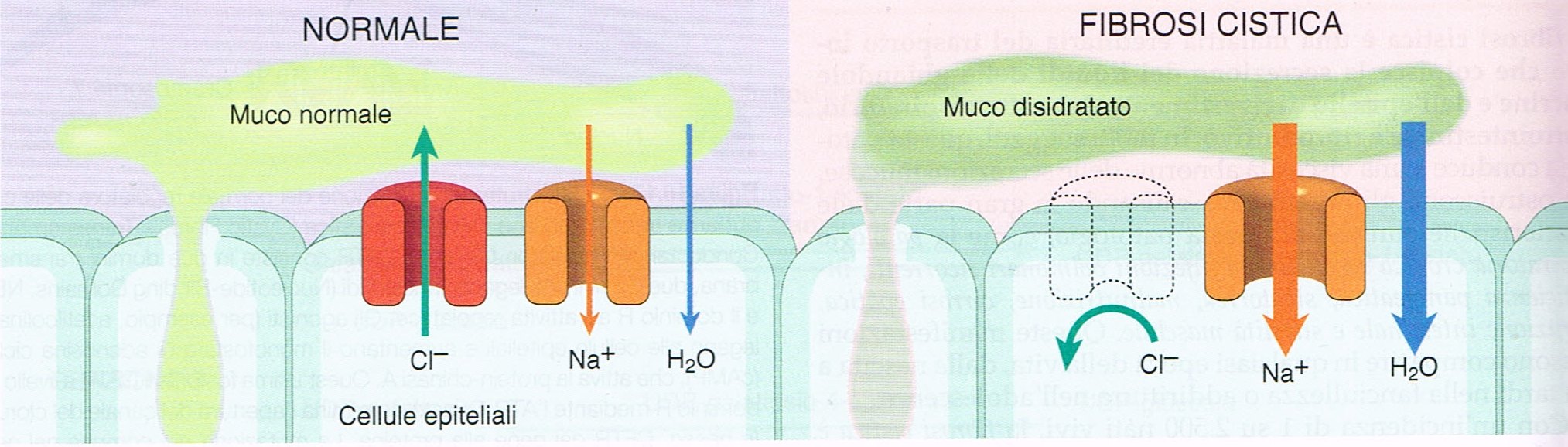
La fibrosi cistica può essere considerata la patologia multisistemica per eccellenza, potendo coinvolgere tutti gli apparati principali del corpo umano. Un effetto devastante che è generato da un unico gene mutato, situato sul cromosoma 7 e codificante per la proteina CFTR, Regolatore della Conduttanza Transmembrana della Fibrosi Cistica, un nome complesso che indica un canale del cloro situato su molte cellule dell’organismo. Il CFTR è fondamentale, in quanto regola soprattutto la composizione delle secrezioni nell’apparato respiratorio e digerente, oltre a determinare la composizione del sudore. Gli affetti da fibrosi cistica, in
virtù di quanto detto, possono presentare una varietà smisurata di sintomi più o meno gravi, tra cui patologie respiratorie con conseguenti infezioni polmonari, malnutrizione, sterilità nei maschi (95% dei casi), insufficienza pancreatica (85-90% dei casi) e steatorrea (grassi indigeriti nelle feci). Tutto ciò avviene perché un malfunzionamento del canale CFTR o una sua ridotta presenza determinano delle secrezioni mucose eccessivamente dense che, ad esempio, portano ad una ostruzione delle vie respiratorie ed a probabili infezioni da stafilococchi, pseudomonas ed altri microrganismi patogeni. Il quadro clinico, tuttavia, è molto variabile perché non tutti gli affetti presentano tutti i sintomi e questi sopraggiungono con tempistiche e gravità differenti.
Una malattia (non troppo) rara
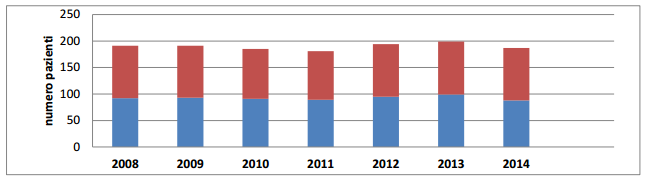
Dal punto di vista genetico la fibrosi cistica è un crogiuolo di mutazioni. Sebbene il gene coinvolto sia uno, le sue possibili mutazioni sono più di 1800 e non tutte sono conosciute e studiate. La maggior parte degli affetti presenta una delezione dell’amminoacido fenilalanina in posizione 508 (ΔF508), che risulta quindi assente e determina un cattivo ripiegamento della proteina-canale e una riduzione della sua emivita. La patologia però si manifesta solo in omozigosi, ovvero solo quando entrambi gli alleli del CFTR sono mutati –anche con mutazioni qualitativamente diverse- nel DNA dell’individuo, che di conseguenza può risultare malato solo se entrambi i genitori sono portatori sani. Questo aspetto della fibrosi cistica è molto importante perché i portatori sani in Italia sono 1/25-26 –un numero elevato-, ma le persone effettivamente malate sono circa circa 1 ogni 2500, con circa 200 nuovi nati all’anno che vengono alla luce con questa patologia. In Campania il centro di riferimento regionale per la FC è parte dell’AOU Federico II di Napoli, dove l’attività è divisa in un’area pediatrica e un’area per il follow up di pazienti adulti. Nell’anno 2014 i pazienti afferenti all’Unità Operativa Semplice pediatrica del centro FC sono stati 187, con 16 nuove diagnosi effettuate.

Diagnosi e cura
In Italia, dato l’elevato numero di portatori sani del gene mutato e considerando che la tempestività della diagnosi è fondamentale per l’aspettativa di vita, è obbligatorio lo screening neonatale, il cui risultato viene poi eventualmente confermato con il test del sudore. Il sudore dei malati di fibrosi cistica è ricco di cloruri (concentrazione maggiore a 60 mEq/L) e di sodio, risultando quindi salato e permettendo ad appositi macchinari di rilevare le concentrazioni elevate di questi elettroliti. Per la fibrosi cistica è disponibile un test genetico, che in una minoranza di casi può rivelarsi problematico, perché non conosciamo tutte le mutazioni del gene in questione e non tutte le mutazioni conosciute hanno una altrettanto chiara correlazione con i vari sintomi. Grazie allo screening ed alle varie opportunità diagnostiche, ad oggi, i casi di fibrosi cistica sono riconosciuti sufficientemente in anticipo per attuare un piano terapeutico multidisciplinare, che ha portato l’aspettativa di vita degli affetti fin quasi ai 40 anni, mentre negli anni ’60 la maggior parte dei malati moriva prima del compimento del quinto anno di età.
Una ricerca made in Italy con un risultato a sorpresa
Fino ad oggi i progressi nelle cure si sono avuti sempre implementando nuovi e migliori farmaci ed approcci terapeutici, improntati a limitare gli effetti deleteri della patologia sui vari apparati colpiti. Un team di ricerca internazionale, guidato da esperti italiani, ha però cambiato la prospettiva, aprendo la strada per una reale cura alla radice della malattia. I ricercatori hanno investigato gli effetti di un polipeptide naturale del nostro organismo, la timosina α1, su topi con una delezione della fenilalanina 508 presente in omozigosi (su entrambi i cromosomi 7) e su cellule dell’epitelio bronchiale umano (HEB) prelevate da cinque soggetti con la stessa delezione. La timosina α1 ha un acclarato effetto immunomodulante ed è utilizzata da tempo come principio attivo dello Zadaxin in patologie come le immunodeficienze. Siccome molti danni nei pazienti affetti da fibrosi cistica si hanno a causa di una infiammazione eccessiva dei tessuti, l’obiettivo del team era quello di verificare se la molecola potesse avere anche in questo ambito un effetto antinfiammatorio. Difatti l’esperimento si è rivelato positivo, con la timosina α1 capace di indurre indirettamente IDO1, una molecola poco espressa nei malati di FC, che modula la risposta immunitaria. Per di più si è notato un effetto non pronosticato della timosina α1, la quale si è mostrata capace di migliorare la localizzazione sulla membrana cellulare e la stabilità del canale CFTR, sia nei topi, migliorandone drasticamente le condizioni di salute, sia nelle cellule di 3 dei 5 soggetti del campione umano. Questo ulteriore effetto potrebbe rappresentare una svolta epocale nel trattamento di una malattia letale come la FC, se i trial clinici dovessero confermarne gli effetti in vivo. Una solida speranza è data dal fatto che la timosina α1, essendo già utilizzata ad altri scopi, è una molecola altamente sicura e già molto studiata, quindi più facilmente gestibile nei trial.


